
In this study, the authors address a relatively new computational challenge: how to to rapidly and accurately identify species-level DNA sequences from next generation metagenomic shotgun sequencing data. NCBI Genbank (v209), the ever-growing library of sequenced DNA fragments mapped to an identified taxonomy species, has an enormous catalog (1.99 x 1011 basepairs of cDNA and genomic DNA from 1.87 x 108 records at the time of this study). Sequence alignment software is used to match unknown DNA sequences to the catalog of identified genetic data, but new approaches are needed to manage the increasing data. In this study, the authors aimed to validate a Scalable Metagenomics Alignment Research Tool (SMART), a novel searching heuristic for shotgun metagenomics sequencing results.
The authors discuss some of the existing techniques for this task, such as Kraken, which works by creating a k-mer database mapped to the lowest common ancestor, reducing the search space significantly. But Kraken requires a long execution time and memory consumption during the database construction, and because the current databases are limited to bacterial, archaeal, and viral genomes, the elimination of host genomic DNA is required prior to classification using Kraken. There are other methods, such as LMAT and CLARK, which have their own advantages and disadvantages.
Recently, the MapReduce programming model caused a substantial shift in the way that large data sets may be distributed in parallel within a computing cluster. In order to avoid limiting the k-mer search space, the authors sought to leverage a MapReduce computational framework with a sharded database and parallel computing to create a scalable complete search heuristic for next generation sequencing files from metagenomics projects.
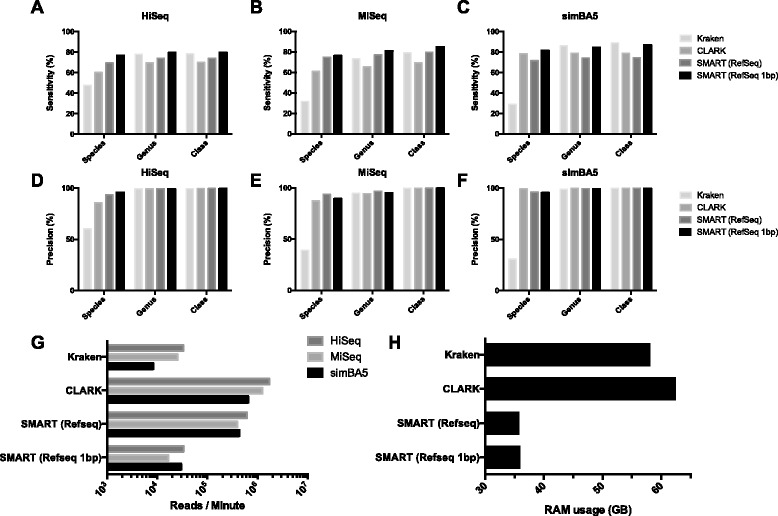
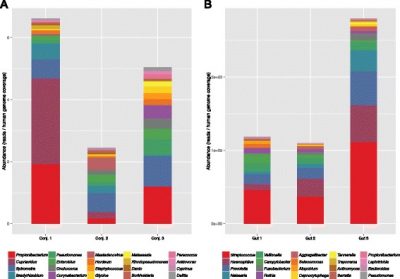
The paper describes the process in detail, but in short, by indexing every 30-mer in the NCBI GenBank with a multiplexed, parallel searching strategy, the authors were able to classify reads against all currently catalogued DNA simultaneously while maintaining similar throughput, sensitivity and precision to Kraken and CLARK on simulated datasets. To the authors' knowledge, this is the first metagenomic classification algorithm capable of efficiently matching against all the species and sequences present in the NCBI GenBank, allowing for a single step classification of microorganisms as well as large plant, mammalian, or invertebrate genomes from which the metagenomic sample may have been derived and allows for identification of novel sequences without pre- or post- filtering steps. This approach will be useful in identifying pathogens, characterizing complex microbiomes, and could be extended to label transcripts in RNASeq data.
Lee AY, Lee CS, Van Gelder RN. Scalable metagenomics alignment research tool (SMART): a scalable, rapid, and complete search heuristic for the classification of metagenomic sequences from complex sequence populations. BMC Bioinformatics. 2016 Jul 28;17:292. doi: 10.1186/s12859-016-1159-6. PubMed PMID: 27465705; PubMed Central PMCID: PMC4963998.